
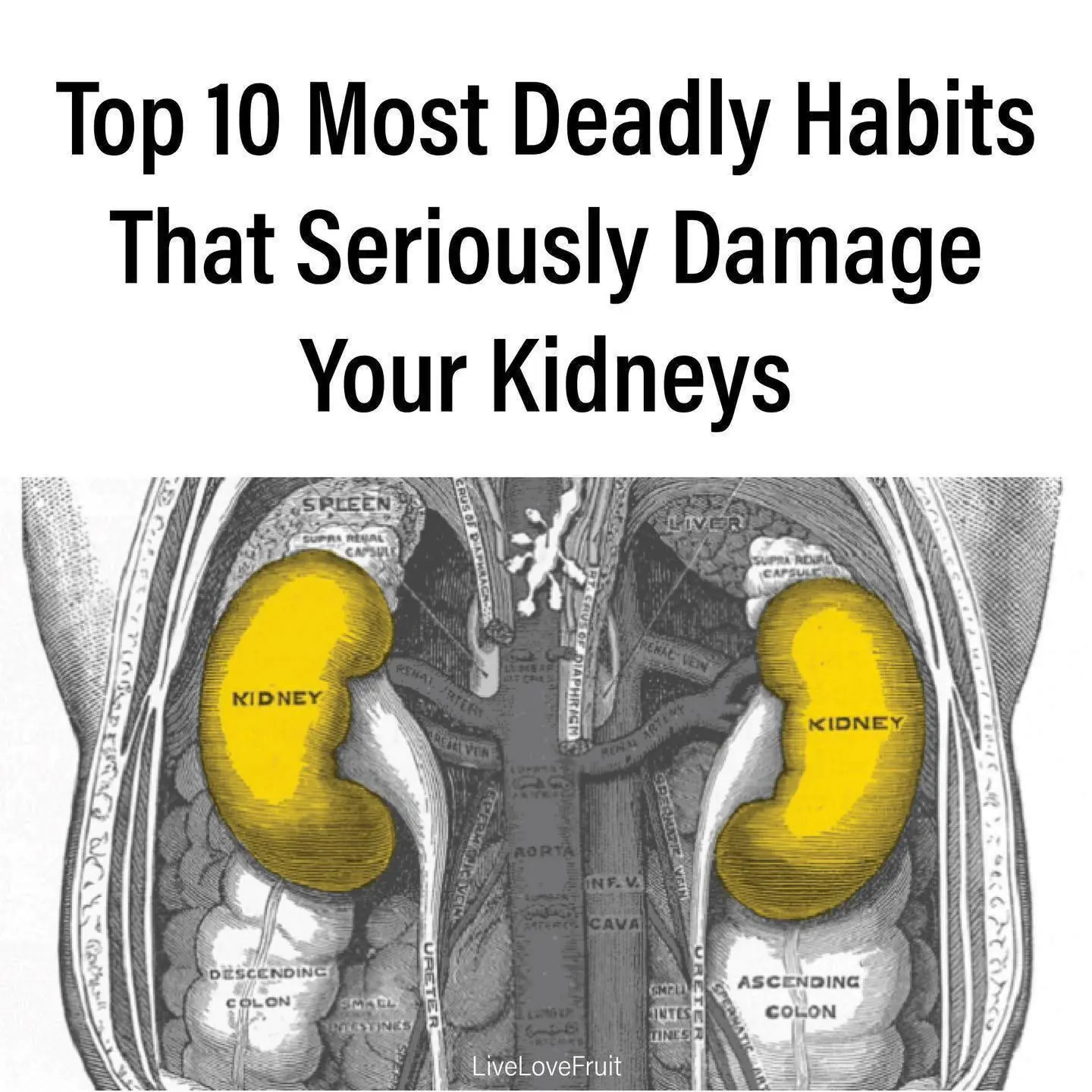
Psoas Muscle Pain Relief: How to Unlock the ‘Muscle of Your Soul’
Health 18/03/2025 02:25

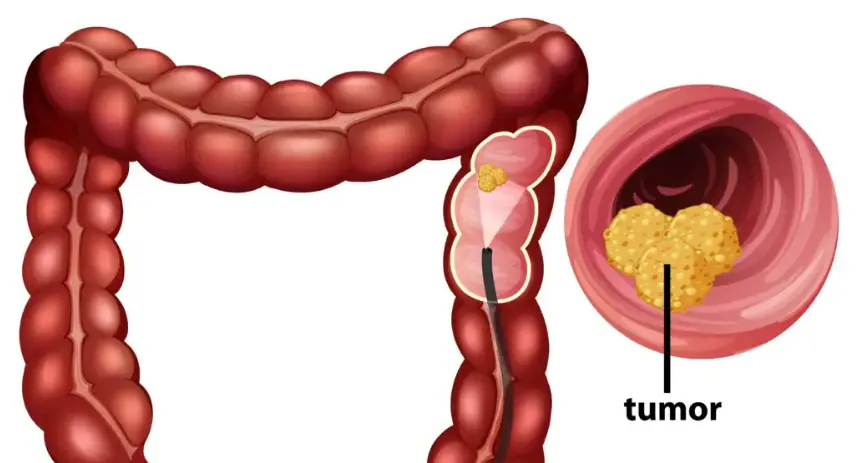




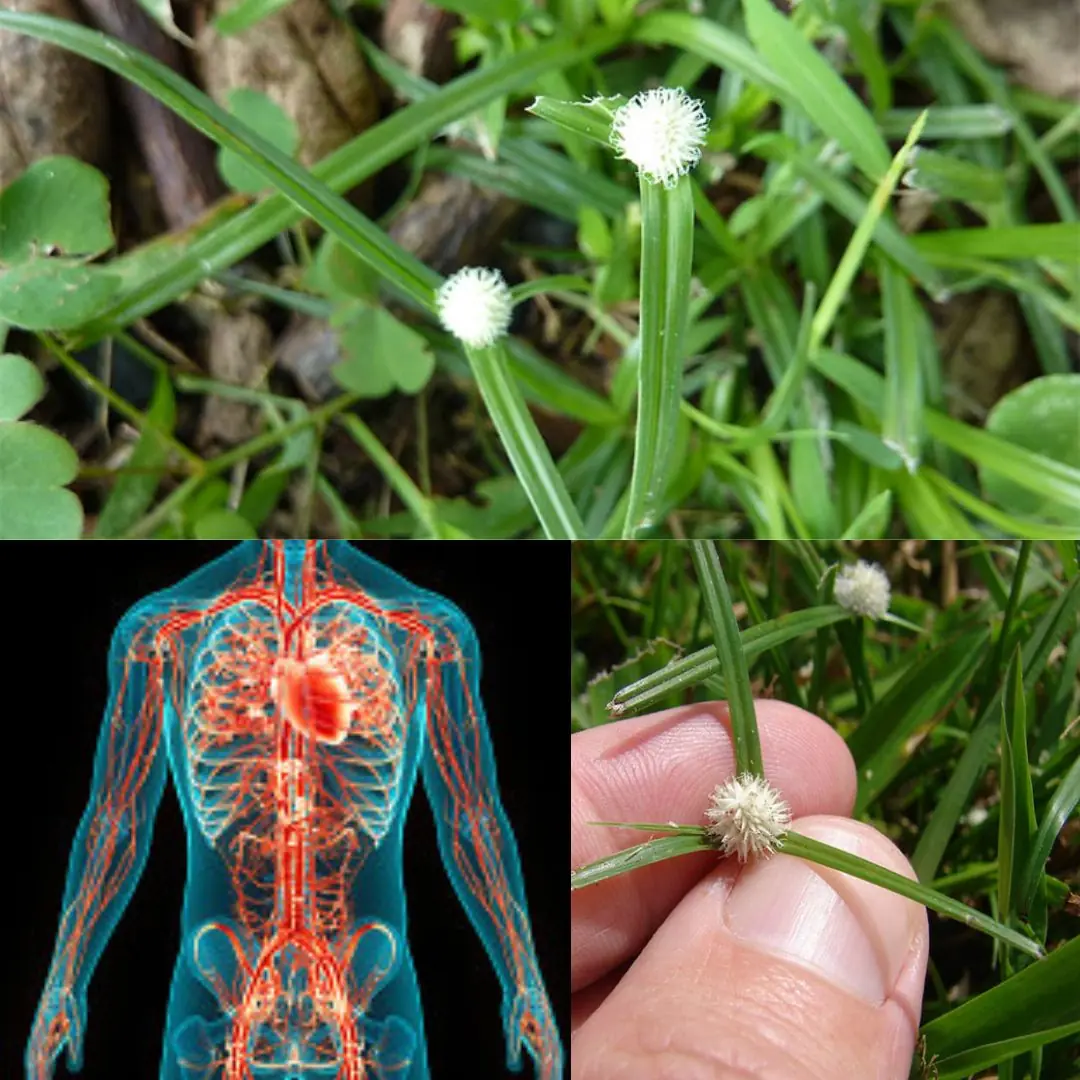







When Elodie's husband, Owen, starts acting distant after the birth of their son, she fears the worst. Sleepless nights and creeping doubts push her to uncover the truth, only to find something she never expected.

When Jessica's husband, James, asks her to be a surrogate for his brother's fiancée, she agrees against her better judgment. Yet, as the pregnancy progresses, her doubts grow. The fiancée remains unreachable, the details feel off, and when Jessica final

When my landlord Amanda tossed my belongings in the trash and locked me out without warning, I thought I had lost everything. But just 24 hours later, I watched her dragging her own furniture to the curb as she faced eviction herself. That was karma. Pure





The rusted chain jutting from the sand seemed worthless to everyone else, but to 13-year-old Adam, it promised an escape from poverty. He couldn't have known that tugging on those corroded links would teach him something far more valuable than gold or sil



When Henry offers shelter to a homeless woman, he doesn't expect much, just a quiet act of kindness. But two days later, his garage is transformed, and Dorothy is nothing like she seemed. As her tragic past unravels, Henry realizes this isn't just about s



